Genialis Server basics
This chapter provides a general overview and explains the basic concepts. We highly recommend reading it even though it is a bit theoretic.
Genialis Server and ReSDK
Genialis Server is a web application that can handle large quantities of biological data, perform complex data analysis, organize results, and automatically document your work in a reproducible fashion. It is based on Resolwe and Resolwe Bioinformatics. Resolwe is an open source dataflow package for the Django framework while Resolwe Bioinformatics is an extension of Resolwe that provides bioinformatics pipelines.
Resolwe SDK for Python allows you to access Genialis Server through Python. It supports accessing, modifying, uploading, downloading and organizing the data.
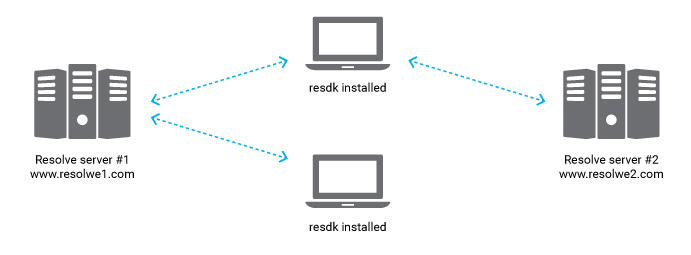
Genialis Server runs on computers with strong computational capabilities. On
the contrary, resdk is a Python package on a local computer that interacts
with Genialis Server through a RESTful API. The power of resdk is its
lightweight character. It is installed with one simple command, but supports
manipulation of large data sets and heavy computation on a remote server.
Data and Process
The two most fundamental resources in Genialis Server are
Data and Process.
Process stores an algorithm that transforms inputs into outputs. It is a blueprint for one step in the analysis.
Data is an instance of a Process. It is a complete record of the performed processing. It remembers the inputs (files, arguments, parameters…), the algorithm used and the outputs (files, images, numbers…). In addition, Data objects store some useful meta data, making it easy to reproduce the dataflow and access information.
Example use case: you have a file reads.fastq with NGS read sequences
and want to map them to the genome genome.fasta with aligner STAR.
Reads are one Data object and genome is another one. Alignment is done by
creating a third Data. At the creation, one always needs to define the Process
(STAR) and inputs (first and second Data). When the Data object is created,
the server automatically runs the given process with provided inputs and
computes all inputs, outputs, and meta data.
Samples and Collections
Eventually, you will have many Data objects and want to organize them. Genialis
server includes different structures to help you group Data objects:
Sample and
Collection.
Sample represents a biological entity. It includes user annotations and Data objects associated with this biological entity. In practice, all Data objects in the Sample are derived from an initial single Data object. Typically, a Sample would contain the following Data: raw reads, preprocessed reads, alignment (bam file), and expressions. A Data object can belong to only one Sample. Two distinct Samples cannot contain the same Data object.
Collection is a group of Samples. In addition to Samples and their Data, Collections may contain Data objects that store other analysis results. Example of this are differential expressions - they are done as combination of many Samples and cannot belong to only one Sample. Each Sample and Data object can only be in one Collection.
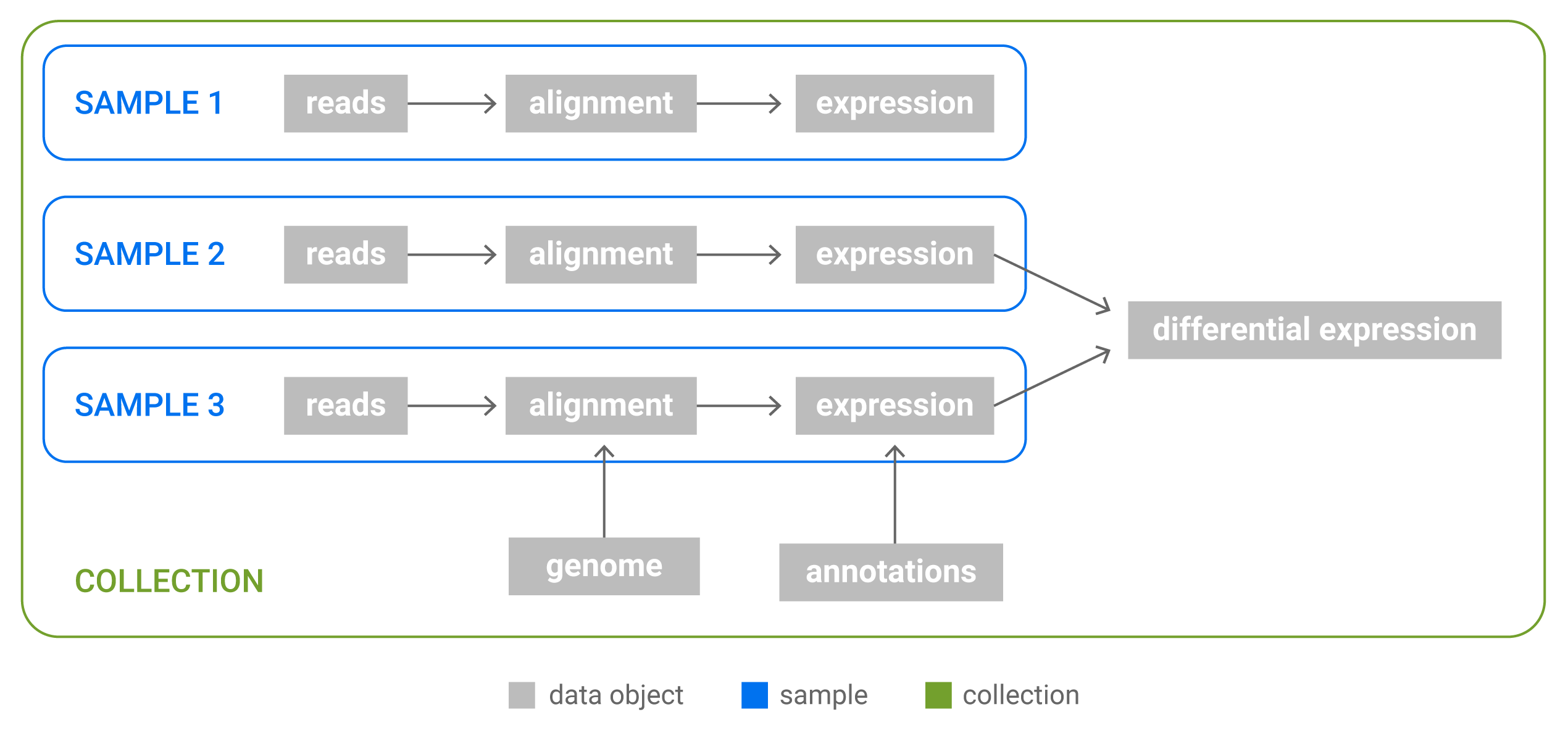
Relations between Data, Samples and Collection. Samples are groups of Data objects originating from the same biological sample: all Data objects in a Sample are derived from a single NGS reads file. Collections are arbitrary groups of Samples and Data objects that store analysis results.
When a new Data object that represents a biological sample (i.e. fastq files, bam files) is uploaded, the unannotated Sample is automatically created. It is the duty of the researcher to properly annotate the Sample. When a Data object that belongs to an existing Sample is used as an input to trigger a new analysis, the output of this process is automatically attached to an existing Sample.